Does cannabidiol reduce the adverse effects of cannabis in schizophrenia? A randomised, do
July 23, 2025
Abstract
In patients with schizophrenia, cannabis use exacerbates symptoms and can lead to a relapse of psychosis. Some experimental studies in healthy volunteers suggest that pre-treatment with cannabidiol (CBD) may reduce these effects, but others do not. Here, we investigated whether pre-treatment with CBD ameliorates the acute adverse effects of cannabis in patients with schizophrenia. Participants (n = 30) had schizophrenia or schizoaffective disorder plus a comorbid cannabis use disorder. In a double-blind, randomised, placebo-controlled, crossover trial, participants received oral CBD 1000 mg or placebo three hours before inhaling vaporised cannabis (containing Δ9-tetrahydrocannabinol (THC) 20–60 mg). The primary outcome was delayed verbal recall measured with the Hopkins Verbal Learning Test-Revised. We also measured psychotic symptoms with the Positive and Negative Syndrome Scale (PANSS) – positive subscale. Delayed verbal recall after cannabis administration was 3.5 words (95% confidence interval [CI]: 2.5–4.5) following pre-treatment with CBD, compared to 4.8 words (95% CI: 3.9 to 5.8) following pre-treatment with placebo (mean difference [MD] = −1.3 [95% CI: −2.0 to −0.6]; p = 0.001). After CBD pre-treatment, inhalation of cannabis was associated with an increase in PANSS-P score of 5.0 (95% CI: 3.6 to 6.5), compared to 2.9 (95% CI: 1.5 to 4.3) following pre-treatment with placebo (MD = 2.2 [95% CI: 0.6 to 3.7]; p = 0.01). Administration of CBD did not have a significant effect on plasma concentration of THC or its active metabolite, 11-hydroxy-THC. In patients with schizophrenia and a comorbid cannabis use disorder, pre-treatment with CBD did not attenuate the acute effects of cannabis on memory impairment or psychotic symptoms, but appeared to exacerbate them. The study was registered on Clinicaltrials.gov (NCT04605393).
Similar content being viewed by others
Introduction
In people with schizophrenia, cannabis use is associated with acute exacerbations of symptoms and an increased risk of relapse [1, 2]. Many people with schizophrenia use cannabis [3], and about one in five have a comorbid cannabis use disorder (CUD) [4]. Managing CUD in people who have schizophrenia is difficult, as there are no specific interventions that are effective in this group [5, 6].
The adverse effects of cannabis are attributable to its constituent Δ9-tetrahydrocannabinol (THC), a partial agonist at the cannabinoid receptor 1 (CB1) [7, 8]. Preclinical data suggest that the effects of THC on memory and behaviour may be mediated by intracellular CB1 receptors which are located on the mitochondria of neurons and astroglia [9, 10]. Neuroimaging studies have suggested that the hippocampus, which is densely populated with CB1 receptors, may mediate many of the behavioural effects of cannabis [11]. When THC is given intravenously to people with schizophrenia, it transiently increases psychotic symptoms and cognitive impairments [12]. In people with both schizophrenia and cannabis use disorder, an oral dose of THC (15 mg) impaired cognition but did not exacerbate psychotic symptoms, while smoked cannabis (THC 3.5%) did not affect either outcome [13]. Another study found that smoked cannabis increased paranoia and impaired cognition in cannabis users at clinical high risk for psychosis, but not in healthy cannabis users [14]. Several controlled studies in healthy volunteers have found that inhaled THC can impair cognition and induce psychotic symptoms when compared to placebo [15, 16].
Cannabidiol (CBD) is another constituent of cannabis and has a similar molecular structure to THC. Its mechanism of action is not established, but it may act as a negative allosteric modulator of CB1 receptors, altering their response to both endogenous agonists, such as anandamide and 2-arachidonoylglycerol, and exogenous agonists, such as THC [17, 18]. Preclinical studies have also suggested that it can modulate mitochondrial function [19,20,21,22], or that its antipsychotic effects may depend on activity at 5-HT1A and TRPV1 receptors [23]. Data from clinical trials suggest that CBD (at doses between 600 and 1000 mg) can reduce psychotic symptoms in people with schizophrenia [24,25,26], and in people at clinical high risk of the disorder [27]. It also has a relatively benign adverse event profile and has a high level of acceptability to people with psychosis [28, 29], and is thus a promising candidate treatment [30].
Several experimental studies in healthy volunteers have investigated whether CBD can modulate the acute effects of cannabis or THC [31]. However, these have produced mixed results. Two studies reported that pre-treatment with CBD (either as a high oral dose or intravenously) attenuated the acute effects of subsequent intravenous THC on psychotic symptoms and memory impairment [32, 33]. In contrast, three studies which co-administered CBD and THC (via the oral route) reported that CBD exacerbated the effects of THC on outcomes such as cognition, anxiety, and subjective level of intoxication [34,35,36]. Finally, three studies that co-administered inhaled CBD and THC found that CBD had no influence on THC’s effects [15, 16, 37]. The differences between studies could be related to differences in whether CBD is given as a pre-treatment or is administered at the same time as THC, the route of administration of CBD and THC (oral/inhaled/intravenous), and the doses of CBD and THC used. For example, when CBD and THC are co-administered orally, CBD may increase the effects of THC by inhibiting hepatic first-pass metabolism [34]. If too low a dose of THC is used it may fail to induce adverse effects, precluding the detection of modulatory effects of CBD [15].
The present study was designed to assess whether pre-treatment with CBD could reduce the acute adverse effects of cannabis in people with schizophrenia. The study was a double-blind, randomised, placebo-controlled, crossover trial conducted in a Clinical Research Facility. To account for drug tolerance, participants who did not experience a significant increase in psychotic symptoms with the standard THC dose (20 mg) were invited to complete additional experiments with higher doses of THC (40 mg and then 60 mg).
We tested the hypotheses that, relative to placebo, pre-treatment with CBD (1000 mg oral) would reduce the severity of:
-
i.
cannabis-induced impairment in delayed verbal recall.
-
ii.
cannabis-induced positive psychotic symptoms.
Methods and materials
Design
A randomised, double-blind, two-arm, crossover laboratory study, conducted between October 2021 and July 2023 at the NIHR Clinical Research Facility at King’s College Hospital, London, UK.
Participants
We recruited individuals receiving secondary mental healthcare from the South London and Maudsley NHS Foundation Trust, London, UK. The inclusion criteria were: age 18-65 years; diagnosis of schizophrenia or schizoaffective disorder (satisfying ICD-10 criteria); clinically stable for at least three months; regular (at least weekly) cannabis use for the past 3 months or more; evidence from either clinicians or from the patient that cannabis use exacerbates their symptoms or increases their risk of relapse (assessed via review of clinical notes, clinician reports, and participant self-report); treatment with regular doses of antipsychotic medication for at least 1 month; participant agrees to abstain from cannabis use for at least 24 hours prior to study visits; willing to have an intravenous cannula inserted to collect blood samples on experimental visits; sufficiently fluent in English; and providing written informed consent. Exclusion criteria were: extreme cannabis use (estimated to use over 2 grams of cannabis/day); dependence on alcohol or illicit substances other than cannabis as defined by ICD-10; pregnancy (current or planned) or breastfeeding; physical health disorder or another mental health disorder that may influence the patient’s ability to tolerate the procedure, or that may alter the results of the study; participant has taken part in any drug study within the last 3 months or taking part in another study over the course of the trial; drug sensitivity/allergy to cannabis or lorazepam; unlikely to be able to complete experiments for any reason.
Screening visit
After providing written informed consent, participants were assessed against inclusion and exclusion criteria, completed a physical health assessment, provided a urine and breath sample for drug and alcohol screening tests, and provided a blood sample to confirm adherence to oral antipsychotic medication. Once these were completed, participants practised the study procedures, i.e. using the vaporiser and completing cognitive and psychopathological assessments.
Randomisation and blinding
The randomisation schedule was developed by an independent statistician and shared with the dispensing pharmacy only. To maintain the balance of treatment allocation, a block randomisation was used, with a block size of four. The study was double-blind, with both participants and researchers blind to treatment allocation. The CBD and placebo capsules had identical appearance.
Participants were asked to guess treatment allocation prior to cannabis intoxication on the first experiment and at the end of the second experiment. Researchers guessed treatment allocation at the end of the second experiment only.
Study drugs
CBD capsules and matching placebo were obtained from BSPG Laboratories, UK. The capsules contained 200 mg of naturally derived CBD which had been refined to >99.9% purity, with no detectable THC, Δ8-THC or cannabinol. The CBD was dissolved in Softisan 378, a medium-chain fatty acid, containing palmitoyl ascorbic acid, which is solid at room temperature.
Two cannabis-based products for medical use (CBPMs) were used during the study: Adven EMT-1 (THC: 19%; CBD < 1%) and Pedanios 20/1 (THC: 18.6–19.8%; CBD < 0.1%), provided by Rokshaw Laboratories, UK and IPS Pharma, UK, respectively.
Drug administration and inhalation procedure
CBD 1000 mg (5 × 200 mg capsules) was administered with food (typically two yoghurts [238–296 kCal; 6.7–8.1 grams of fat]).
For the cannabis challenge, ground cannabis flower, containing THC 20 mg (four standard THC units [38]), was vaporised and then inhaled by participants. Cannabis inhalation began three hours after CBD administration (aligning with CBD’s expected peak plasma concentration [39]) and followed the method used by Englund and colleagues [37]. The cannabis was vaporised at 210 °C into a transparent polythene balloon using a Volcano® Medic Vaporiser (Storz-Bickel GmbH, Tüttlingen, Germany). Participants were instructed to hold their breath for 8 seconds after each inhalation, with an 8 second break between inhalations. After a short break (1–2 minutes), the procedure was repeated, and the cannabis was heated again to inflate a second balloon.
Dose-escalation procedure
To take into account drug tolerance, and to increase the power of the study, participants who did not demonstrate a significant psychotic response to THC 20 mg (defined as an increase in PANSS-positive subscale of ≥3 during either experiment) [32, 37, 40] were invited to complete additional pairs of experiments at higher doses (Figure S1). The higher doses were 40 mg (eight standard THC units) and then 60 mg (12 standard THC units). Participants were re-randomised.
Experimental procedure
At the start of each experiment, participants provided a urine sample to confirm recent abstinence from substances (apart from cannabis), completed a breath test to confirm recent abstinence from alcohol, and were judged as being sober by the study psychiatrist. Psychopathological outcome measures were completed prior to CBD/placebo administration and were then repeated at later timepoints (Figure S2). Cognitive testing was completed once during each experiment, starting 20 minutes after cannabis inhalation was completed. Caffeinated drinks and cigarette breaks were allowed ad libitum. At the end of the study visit, participants were discharged after completing a sobriety test.
Outcome measures
Hopkins verbal learning test-revised (HVLT-R)
The primary outcome measure was delayed verbal recall on the HVLT-R [41]. A review of the effects of THC on cognition found that some of the largest impairments occur on tasks measuring verbal learning and memory [42]. The HVLT-R was completed at screening and once during each experiment, 20 minutes after cannabis intoxication. The researcher reads a list of 12 words to the participant and asks them to repeat aloud as many words as they can remember. This has been completed three times. The total number of words recalled across the three learning trials was the immediate recall score. After 20–25 minutes, participants are asked to recall the list of words, to determine delayed recall. An intrusion was a word which was not in the list. Repetitions referred to the number of times a correctly recalled word was repeated. A unique list of words was used on each study visit.
The positive and negative syndrome scale (PANSS) – positive and negative subscales
The Positive and Negative Syndrome Scale is the gold-standard assessment for measuring symptom severity in schizophrenia [43]. We assessed the Positive and Negative subscales. The Positive Subscale (PANSS-P) was the psychopathological outcome of primary interest. A semi-structured interview was conducted by a psychiatrist trained in PANSS assessment and was complemented by observations from other researchers. The PANSS is typically used to assess psychotic symptoms over the past week. In the present study, the PANSS was used to measure acute symptom changes, an approach that has been used in several previous experimental cannabis studies [12, 13, 16, 32, 37]. The initial interview assessment was completed prior to CBD/placebo administration. The post-THC assessment included an interview at 0–20 minutes after inhalation and a second interview/review at the end of the experiment.
The psychotomimetic states inventory (PSI)
The PSI was used to assess subjective psychotic symptoms [44]. The questionnaire includes 48 items organised into six domains: delusory thinking, perceptual distortion, cognitive disorganization, anhedonia, mania, and paranoia. The PSI was completed prior to CBD/placebo administration, and at least 90 minutes after cannabis administration. Participants were instructed to describe their experiences over the last few hours, or since receiving cannabis.
The state social paranoia scale (SSPS)
The SSPS is a ten-item questionnaire to assess persecutory thoughts [45]. The SSPS was completed prior to CBD/placebo administration, and at least 90 minutes after cannabis administration. Participants were instructed to describe their experiences over the last few hours, or since receiving cannabis.
The state-trait anxiety inventory-state scale (STAI-S)
The STAI-S is a 20-item scale for assessing anxiety symptoms [46]. The STAI-S was completed at three timepoints: prior to CBD/placebo administration, immediately prior to cannabis administration, and 20 minutes after cannabis administration.
Forward and reverse digit span
This measure of working memory involves the recall of increasingly long sequences of numbers in forward and reverse order. The task continues until the participant fails two consecutive attempts at a sequence. It was completed 25 minutes after cannabis administration.
Visual analogue scales (VAS)
These were used to measure other subjective effects. Participants marked on a 100 mm horizontal line to indicate the level of a given feeling at that moment (0 mm ‘Not at all’ to 100 mm ‘Extremely’). The feeling states included: ‘Feel drug effect’, ‘Like drug effect’, ‘Want more drug’, ‘Thinking clearly’, ‘Tired’, ‘Excited’, ‘Want to talk’, ‘Anxious’, ‘Relaxed’, ‘Happy,’ ‘Irritable’, ‘Suspicious’, ‘Hearing voices’, ‘Dry mouth’, ‘Hungry’, ‘Vulnerable’, and ‘Threatened’. VAS were complete at seven timepoints: baseline, 90 minutes post-CBD/placebo, immediately prior to cannabis administration, 10-, 45- and 90-minutes post-cannabis, and at the end of the study visit.
Pharmacokinetics
Blood samples were collected at six timepoints: pre-CBD/placebo administration, 90 minutes post-CBD/placebo, and at 0-, 5-, 15- and 90-minutes post-cannabis inhalation. The concentration of cannabinoids was quantified using ultra high-performance liquid chromatography-mass spectrometry.
Physiological outcomes
Heart rate, blood pressure and temperature were measured prior to CBD/placebo administration, 90 minutes post-CBD/placebo, at 5-, 15- and 90-minutes post-cannabis inhalation, and at discharge.
Statistical analysis
Registration
The study was registered on Clinicaltrials.gov (NCT04605393) and Open Science Framework (https://osf.io/2y4n8/). The statistical analysis plan was registered prior to unblinding of treatment allocation (https://osf.io/xswdm).
Primary analysis population
Since this is an experimental study, which aims to understand the efficacy of CBD under ideal conditions, not its effectiveness as a clinical intervention, our primary analyses were per-protocol. For participants who completed more than one pair of experiments, only the data from the experiments where they received the highest dose of THC were included.
Sensitivity analyses
We completed two sensitivity analyses for delayed verbal recall and change in PANSS-P: i) with the intention to treat population (highest dose received) (n = 34), and ii) using data from THC 20 mg experiments only (n = 30).
Analysis of psychopathology, cognition and physiology
Continuous outcomes are reported as means with standard deviation (SD). Categorical outcomes are reported as frequencies. We used linear mixed models to assess the effect of CBD on continuous outcomes. Treatment allocation (CBD/placebo) and visit number were included as fixed effects, with participant as a random effect to account for the dependency between repeated measures. Estimated marginal means were calculated and compared in a pairwise test. Treatment effects on rate of clinically significant PANSS-P increases (≥3) were assessed with McNemar’s test.
Pharmacokinetic analyses
Pharmacokinetic parameters were summarized descriptively. We reported the geometric mean of the peak plasma concentration (Cmax) and plasma exposure over time (AUCt). AUCt was calculated using the trapezoid method implemented in the bayestestR package (version 0.13.0). To compare the Cmax and AUCt of pharmacokinetic parameters we followed the same analysis strategy as for psychopathological and cognitive outcomes.
Imputation of data
There was no missing data for cognitive and psychopathological outcomes. Multiple imputation chain equations (MICE) were used to impute missing values in pharmacokinetic outcomes using the mice package (version 3.16.0) in R. A total of 37/360 (10%) data points were missing.
Order effects
There was sufficient time for drug washout between experiments (minimum 1 week), so carryover effects were considered unlikely, but due to learning and familiarisation with the study protocol and lab environment, a period effect may have occurred. We therefore included visit number in all models to account for potential order effects.
Correction for multiple comparisons
Benjamini-Hochberg correction was conducted across outcomes locally i.e. across secondary cognitive outcomes, secondary psychopathological outcomes, VAS, and physiological outcomes. P values below 0.05 were considered significant. All tests were two-tailed.
Software
All analyses were conducted using R version 4.2.2. lme4 (version 1.1-31) was used to fit the linear mixed effects models and estimated marginal mean (EMM) contrasts were calculated using the emmeans package (version 1.8.4-1).
Ethical and regulatory approvals
The study was approved by the Health and Social Care Research Ethics Committee A (Reference Number: 20/NI/0074). All participants provided written informed consent and the study was conducted in compliance with the principles of Good Clinical Practice, and the Declaration of Helsinki (1996).
Results
Recruitment, pilot experiments and dose-escalation
273 individuals were screened for inclusion, of which 92 were invited to an assessment (Figure S3). Of these, 43 attended the assessment and 37 completed at least one experiment. Three participants completed pilot experiments with low doses of THC (10 mg and 15 mg) only, and four participants dropped out after their first experiment.
Thirty participants finished the study per-protocol, each having completed two experiments with THC 20 mg. Of these, 19 had a significant response to the cannabis challenge (PANSS-P increase ≥3). Eleven participants were eligible to repeat the experiments with THC 40 mg of which six volunteered. One participant did not respond to THC 40 mg, so they completed two experiments with THC 60 mg. Five participants did not complete higher doses due to: loss to follow-up (n = 2), alcohol use disorder relapse, death, and declined.
Reasons for withdrawal and adverse events
Four participants dropped out of the study after their first experimental visit (Figure S3). One withdrew due to a newly diagnosed medical condition, unrelated to the study procedures. Another discontinued treatment with antipsychotic medication prior to their second experiment. One participant disagreed with the timing of blood sampling and therefore withdrew.
One participant was withdrawn due to a treatment-related adverse event. During their first experiment, soon after receiving cannabis, they developed persecutory delusions related to the study team. By the following day, and without additional intervention, these symptoms resolved completely. One other participant experienced a treatment-related adverse event. After receiving the cannabis, their blood pressure reached 224/78 mm Hg. By the end of the experiment it had normalised without intervention. They did not withdraw from the study as it occurred during their second experiment. Both treatment-related adverse events occurred after treatment with CBD.
Per-protocol analysis
The demographic and clinical characteristics of the per-protocol study population are described in Table 1. The characteristics of the intention-to-treat and higher dose study populations are reported in Table S1. There was no difference in the time taken to complete the cannabis administration procedure between the two arms (CBD: 11.8 minutes; Placebo: 11.5 minutes; p = 0.76).
The primary outcome was delayed verbal recall after cannabis administration (Table 2, Fig. 1A). Lower scores indicate worse performance. In the CBD arm, the mean score was 3.5 (95% CI: 2.5 to 4.5); in the placebo arm, it was 4.8 (95% CI: 3.9 to 5.8), a difference that was statistically significant (MD = -1.3 [95% CI: -2.0 to -0.6]; p = 0.001). There was no effect of visit (p > 0.05). Sensitivity analyses produced the same pattern of results (Tables S2 and S3).
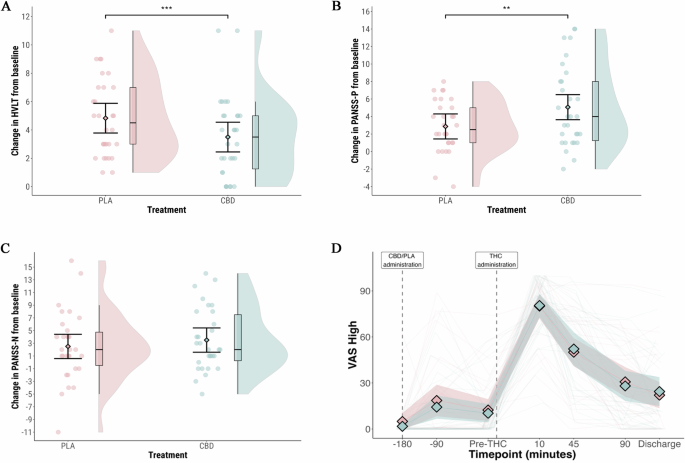
A Delayed verbal learning on the HVLT-R; B Positive psychosis symptoms measured by the PANSS-Positive Subscale; C Negative psychosis symptoms measured by the PANSS-Negative Subscale; D Participant rated level of intoxication measured with a visual analogue scale for ‘Feel Drug Effect’. Blue: CBD pre-treatment arm; Pink: Placebo pre-treatment arm. Shaded areas in violin plots (1A-C) represent distribution density. In Fig. 1D, the shaded areas represent 95% confidence intervals. **p = 0.01 ***p = 0.001.
PANSS-P was assessed at baseline (before CBD/placebo treatment) and after cannabis (Table 3, Fig. 1B). In the CBD arm, the mean increase was 5.0 (95% confidence interval [CI]: 3.6 to 6.5); in the placebo arm, it was 2.9 (95% CI: 1.5 to 4.3). The difference in effect between the two arms was statistically significant (estimated marginal mean difference [MD] = 2.2 [95% CI: 0.6 to 3.7]; p = 0.01). A large increase in PANSS-P (increase of ≥9) was observed in seven participants in the CBD arm, and in no participants in the placebo arm (p = 0.000005). There was no effect of visit (p > 0.05). Sensitivity analyses produced the same pattern of results (Tables S4 and S5).
Out of the seven items on the PANSS-P, two had statistically significant differences between treatment arms. For conceptual disorganisation, the mean increase in the CBD arm was 2.2 (95% CI: 1.7 to 2.6); in the placebo arm, it was 1.5 (95% CI: 1.1 to 1.9) (MD = 0.7 [95% CI: 0.2 to 1.1]; p = 0.01; corrected p = 0.15). For suspiciousness/persecution, in the CBD arm, the mean increase was 0.8 (95% CI: 0.4 to 1.2); in the placebo arm, it was 0.2 (95% CI: −0.1 to 0.6) (MD = 0.6 [95% CI: 0.1 to 1.0]; p = 0.01; corrected p = 0.15).
CBD pre-treatment was associated with greater impairment of immediate recall on the HVLT-R (p = 0.04), but this did not survive correction for multiple comparisons (p = 0.14) (Table 2). There was no effect of CBD treatment on participant rated level of intoxication, measured with a VAS for ‘Feel drug effect’ (Fig. 1D), the mean increase was 78.7 (95% CI: 69.0 to 88.4) in the CBD arm, and 75.2 (95% CI: 65.5 to 84.9) in the placebo arm (p = 0.42). There was no effect of CBD treatment on negative symptoms (Fig. 1C), other cognitive or psychopathological outcomes (Tables 2 and 3); or VAS (Table S6). Physiological outcome measures are presented in Table S7. There was a greater increase in systolic blood pressure in the CBD treatment group between baseline and peak (MD = 11.2 mm Hg [3.7 to 18.6]; p = 0.01; corrected p = 0.04).
Assessment of blinding
Participant blinding was assessed on two occasions. During the first experiment, immediately prior to THC challenge, participants correctly guessed their treatment allocation on 16/30 (53%) of occasions. At the end of the second experiment, participants correctly guessed their treatment allocation on 14/30 (47%) occasions. Researcher blinding was assessed at the second timepoint. They correctly guessed treatment allocation on 5/30 (17%) of occasions (James’ Blinding Index = 0.76 [95% CI: 0.58 to 0.94]).
Pharmacokinetics
The pharmacokinetic parameters of THC, CBD and their metabolites are presented in Table S8. We found no evidence for an effect of CBD treatment on the plasma exposure of THC, or its active metabolite 11-hydroxy-THC. CBD treatment was associated with increased plasma exposure of 11-carboxy-THC. The plasma concentration-time profile of THC and CBD are presented in Fig. 2A-D. There was a statistically significant correlation between CBD AUC and change in PANSS-P (r = 0.43 [0.19 to 0.61], p = 0.018) and HVLT delayed recall (r = −0.27 [−0.49 to −0.02], p = 0.038). There was no significant correlation between the plasma concentration of THC or 11-hydroxy-THC and delayed verbal recall or change in PANSS-P (Table S9 and Fig. S4).
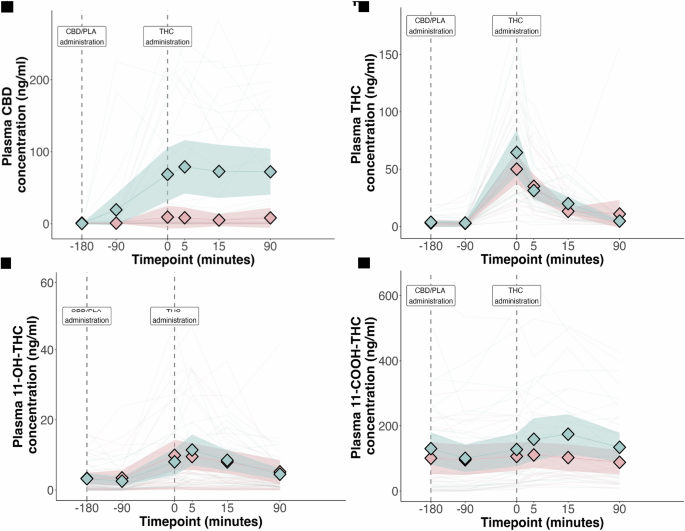
Plasma concentration of (A) CBD; (B) THC; (C) 11-hydroxy-THC; and (D) 11-carboxy-THC. Data presented are arithmetic mean with 95% CI. Blue: CBD pre-treatment arm; Pink: Placebo pre-treatment arm. Shaded areas represent the 95% confidence interval.
Discussion
To our knowledge, this is the first study to investigate if CBD can moderate the acute effects of cannabis in people with either schizophrenia or a cannabis use disorder. Our main finding was that CBD increased severity of verbal memory impairment and positive psychotic symptoms following cannabis administration. Our findings were not attributable to a pharmacokinetic interaction between CBD and THC.
Some previous studies in healthy volunteers have found that CBD increases the acute adverse effects of THC when they are co-administered orally [34,35,36]. However, this is because CBD inhibits the hepatic metabolism of THC through the inhibition of CYP enzymes [34]. In the present study, pharmacokinetic analyses indicated that CBD pre-treatment did not affect the plasma exposure of THC or its pharmacologically active metabolite, 11-hydroxy-THC. Furthermore, in a post-hoc analysis, there was a significant correlation between CBD plasma concentration and positive psychotic symptoms and memory impairment, correlations that were not evident with either THC or 11-hydroxy-THC. These findings suggest that the exacerbation of THC’s effects by CBD was unlikely to be the result of a pharmacokinetic mechanism, and may have been due to a pharmacodynamic effect.
Our results contrast with data from two studies in healthy volunteers which reported that pre-treatment with CBD reduced the adverse effects of THC [32, 33]. However, both schizophrenia and cannabis use disorder are associated with significant alterations in the brain endocannabinoid system [11, 47, 48], as well as mitochondrial function [49, 50], which could modify the effects of CBD and THC. In the present study, we chose to examine a subgroup of patients with schizophrenia who also had a CUD, because we were interested in assessing the potential of CBD as a treatment for patients with schizophrenia whose symptoms are exacerbated by cannabis use. As a result, it is unclear whether the differences between the present findings and those previously reported in healthy volunteers are related to an effect of schizophrenia, of CUD, or both. Most patients with schizophrenia do not have CUD [4], and future studies could investigate whether the effects we observed are also evident in patients who don’t use cannabis, or only do so occasionally. Similarly, it would be useful to repeat our investigation in people who have a CUD but do not have schizophrenia.
CBD is a candidate novel treatment for schizophrenia: a number of trials suggest that it can reduce psychotic symptoms in patients with the disorder, and in people at clinical high risk for the disorder [24, 25, 27]. It is important to emphasise that the results of the present study relate to effects of CBD on psychotic symptoms experimentally induced by THC, rather than symptoms of schizophrenia itself. Moreover, our study involved participants with both schizophrenia and a CUD, as opposed to schizophrenia alone. Clinical trials investigating CBD as a treatment for schizophrenia should compare its efficacy and safety in individuals with and without comorbid CUD.
Strengths of our study include the use of a high oral dose of CBD, equivalent to the oral dose used in clinical trials of schizophrenia, and the administration of THC via inhalation, the route employed by most recreational cannabis users. The incorporation of a THC dose-ranging approach reduced the proportion of participants who did not show a symptomatic response to cannabis, increasing our ability to detect a modulatory effect of CBD. One limitation is that participants were administered a fixed dose of THC, rather than titrating their intake according to their desired level of intoxication. It is therefore unclear whether the effects associated with CBD in this study would occur in a real-world setting. Other limitations are that we only investigated a single dose of CBD and that the sample was predominantly male. Finally, the crossover design, pre-registered statistical analysis plan, and collection of pharmacokinetic data strengthen the internal validity of the study.
Our findings do not support the use of CBD as a means of ameliorating the acute adverse effects of cannabis in people with schizophrenia. However, because we studied patients with comorbid CUD, we cannot exclude the possibility that CBD might reduce the effects of cannabis in patients with schizophrenia who do not have a comorbid CUD.
References
-
Henquet C, Van Os J, Kuepper R, Delespaul P, Smits M, Campo JÀ, et al. Psychosis reactivity to cannabis use in daily life: An experience sampling study. Br J Psychiatry. 2010;196:447–53.
-
Schoeler T, Monk A, Sami MB, Klamerus E, Foglia E, Brown R, et al. Continued versus discontinued cannabis use in patients with psychosis: A systematic review and meta-analysis. Lancet Psychiatry. 2016;3:215–25.
-
Di Forti M, Quattrone D, Freeman TP, Tripoli G, Gayer-Anderson C, Quigley H, et al. The contribution of cannabis use to variation in the incidence of psychotic disorder across Europe (EU-GEI): a multicentre case-control study. Lancet Psychiatry. 2019;6:427–36.
-
Hunt GE, Large MM, Cleary M, Lai HMX, Saunders JB. Prevalence of comorbid substance use in schizophrenia spectrum disorders in community and clinical settings, 1990–2017: systematic review and meta-analysis. Drug Alcohol Depend. 2018;191:234–58.
-
Lees R, Hines LA, D’Souza DC, Stothart G, DI Forti M, Hoch E, et al. Psychosocial and pharmacological treatments for cannabis use disorder and mental health comorbidities: A narrative review. Psychol Med. 2021;51:353–64.
-
Rogers J, Chesney E, Morrison P, Gaughran F Drug Treatment of the Psychoses. Seminars in General Adult Psychiatry, Cambridge University Press; 2024. p. 241.
-
Morrison PD, Zois V, McKeown DA, Lee TD, Holt DW, Powell JF, et al. The acute effects of synthetic intravenous [Delta] 9-tetrahydrocannabinol on psychosis, mood and cognitive functioning. Psychol Med. 2009;39:1607.
-
Oliver D, Chesney E, Cullen AE, Davies C, Englund A, Gifford G, et al. Exploring causal mechanisms of psychosis risk. Neurosci Biobehav Rev. 2024;162:105699.
-
Hebert-Chatelain E, Desprez T, Serrat R, Bellocchio L, Soria-Gomez E, Busquets-Garcia A, et al. A cannabinoid link between mitochondria and memory. Nature. 2016;539:555–9.
-
Jimenez-Blasco D, Busquets-Garcia A, Hebert-Chatelain E, Serrat R, Vicente-Gutierrez C, Ioannidou C, et al. Glucose metabolism links astroglial mitochondria to cannabinoid effects. Nature. 2020;583:603–8.
-
Bloomfield MAP, Hindocha C, Green SF, Wall MB, Lees R, Petrilli K, et al. The neuropsychopharmacology of cannabis: A review of human imaging studies. Pharm Ther. 2019;195:132–61.
-
D’Souza DC, Perry E, MacDougall L, Ammerman Y, Cooper T, Braley G, et al. The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: implications for psychosis. Neuropsychopharmacology. 2004;29:1558–72.
-
Brunette MF, Roth RM, Trask C, Khokhar JY, Ford JC, Park SH, et al. Randomized laboratory study of single-dose cannabis, dronabinol, and placebo in patients with schizophrenia and cannabis use disorder. Schizophr Bull. 2024;51:479–92.
-
Vadhan NP, Corcoran CM, Bedi G, Keilp JG, Haney M. Acute effects of smoked marijuana in marijuana smokers at clinical high-risk for psychosis: A preliminary study. Psychiatry Res. 2017;257:372–4.
-
Morgan CJA, Freeman TP, Hindocha C, Schafer G, Gardner C, Curran HV. Individual and combined effects of acute delta-9-tetrahydrocannabinol and cannabidiol on psychotomimetic symptoms and memory function. Transl Psychiatry. 2018;8:1–10.
-
Lawn W, Trinci K, Mokrysz C, Borissova A, Ofori S, Petrilli K, et al. The acute effects of cannabis with and without cannabidiol in adults and adolescents: A randomised, double-blind, placebo-controlled, crossover experiment. Addiction. 2023;118:1282–94.
-
Laprairie RB, Bagher AM, Kelly MEM, Denovan-Wright EM. Cannabidiol is a negative allosteric modulator of the cannabinoid CB1 receptor. Br J Pharm. 2015;172:4790–805.
-
Navarro G, Reyes-Resina I, Rivas-Santisteban R, de Medina VS, Morales P, Casano S, et al. Cannabidiol skews biased agonism at cannabinoid CB1 and CB2 receptors with smaller effect in CB1-CB2 heteroreceptor complexes. Biochem Pharm. 2018;157:148–58.
-
Drummond-Main CD, Ahn Y, Kesler M, Gavrilovici C, Kim DY, Kiroski I, et al. Cannabidiol impairs brain mitochondrial metabolism and neuronal integrity. Cannabis Cannabinoid Res. 2023;8:283–98.
-
Fišar Z, Singh N, Hroudová J. Cannabinoid-induced changes in respiration of brain mitochondria. Toxicol Lett. 2014;231:62–71.
-
Ryan D, Drysdale AJ, Lafourcade C, Pertwee RG, Platt B. Cannabidiol targets mitochondria to regulate intracellular Ca2+ levels. J Neurosci. 2009;29:2053–63.
-
Nunn AVW, Guy GW, Bell JD. Informing the cannabis conjecture: from life’s beginnings to mitochondria, membranes and the electrome—a review. Int J Mol Sci. 2023;24:13070.
-
Pedrazzi JFC, Silva-Amaral D, Issy AC, Gomes FV, Crippa JA, Guimarães FS, et al. Cannabidiol attenuates prepulse inhibition disruption by facilitating TRPV1 and 5-HT1A receptor-mediated neurotransmission. Pharm Biochem Behav. 2024;245:173879.
-
McGuire P, Robson P, Cubala WJ, Vasile D, Morrison PD, Barron R, et al. Cannabidiol (CBD) as an adjunctive therapy in schizophrenia: A multicenter randomized controlled trial. Am J Psychiatry. 2018;175:225–31.
-
Leweke FM, Piomelli D, Pahlisch F, Muhl D, Gerth CW, Hoyer C, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry. 2012;2:e94–e94.
-
Boggs DL, Surti T, Gupta A, Gupta S, Niciu M, Pittman B, et al. The effects of cannabidiol (CBD) on cognition and symptoms in outpatients with chronic schizophrenia a randomized placebo controlled trial. Psychopharmacol (Berl). 2018;235:1923–32.
-
Bhattacharyya S, Appiah�Kusi E, Wilson R, O’Neill A, Brammer M, Williams S, et al. Effects of cannabidiol on symptoms in people at clinical high risk for psychosis. World Psychiatry. 2024;23:451–2.
-
Chesney E, Lamper D, Lloyd M, Oliver D, Hird E, McGuire P. Acceptability of cannabidiol in patients with psychosis. Ther Adv Psychopharmacol. 2022;12:20451253221128444.
-
Chesney E, Oliver D, Green A, Sovi S, Wilson J, Englund A, et al. Adverse effects of cannabidiol: a systematic review and meta-analysis of randomized clinical trials. Neuropsychopharmacology. 2020;45:1799–806.
-
Chesney E, Oliver D, McGuire P. Cannabidiol (CBD) as a novel treatment in the early phases of psychosis. Psychopharmacol (Berl). 2022;239:1179–90.
-
Freeman AM, Petrilli K, Lees R, Hindocha C, Mokrysz C, Curran HV, et al. How does cannabidiol (CBD) influence the acute effects of delta-9-tetrahydrocannabinol (THC) in humans? A systematic review. Neurosci Biobehav Rev. 2019;107:696–712.
-
Englund A, Morrison PD, Nottage J, Hague D, Kane F, Bonaccorso S, et al. Cannabidiol inhibits THC-elicited paranoid symptoms and hippocampal-dependent memory impairment. J Psychopharmacol. 2013;27:19–27.
-
Bhattacharyya S, Morrison PD, Fusar-Poli P, Martin-Santos R, Borgwardt S, Winton-Brown T, et al. Opposite effects of δ-9-tetrahydrocannabinol and cannabidiol on human brain function and psychopathology. Neuropsychopharmacology. 2010;35:764–74.
-
Zamarripa CA, Spindle TR, Surujunarain R, Weerts EM, Bansal S, Unadkat JD, et al. Assessment of orally administered Δ9-tetrahydrocannabinol when coadministered with cannabidiol on Δ9-tetrahydrocannabinol pharmacokinetics and pharmacodynamics in healthy adults: a randomized clinical trial. JAMA Netw Open. 2023;6:e2254752.
-
Leweke FM, Mueller JK, Reuter AR, Lange B, Pahlisch F, Boost C, et al. Cannabidiol effects on delta-9-tetrahydrocannabidiol-induced psychotic symptoms are related to serum levels of both compounds in healthy volunteers. Neuropsychopharmacology. 2019;44:176.
-
Gorbenko AA, Heuberger JAAC, Klumpers LE, de Kam ML, Strugala PK, de Visser SJ, et al. Cannabidiol increases psychotropic effects and plasma concentrations of Δ9-tetrahydrocannabinol without improving its analgesic properties. Clin Pharmacol Ther. 2024;116:1289–1303.
-
Englund A, Oliver D, Chesney E, Chester L, Wilson J, Sovi S, et al. Does cannabidiol make cannabis safer? A randomised, double-blind, cross-over trial of cannabis with four different CBD:THC ratios. Neuropsychopharmacology. 2023;48:869–76.
-
Freeman TP, Lorenzetti V. A standard THC unit for reporting of health research on cannabis and cannabinoids. Lancet Psychiatry. 2021;8:944–6.
-
Millar SA, Stone NL, Yates AS, O’Sullivan SE. A systematic review on the pharmacokinetics of cannabidiol in humans. Front Pharm. 2018;9:1365.
-
D’Souza DC, Abi-Saab WM, Madonick S, Forselius-Bielen K, Doersch A, Braley G, et al. Delta-9-tetrahydrocannabinol effects in schizophrenia: Implications for cognition, psychosis, and addiction. Biol Psychiatry. 2005;57:594–608.
-
Brandt J. The Hopkins Verbal Learning Test: Development of a new memory test with six equivalent forms. Clin Neuropsychologist. 1991;5:125–42.
-
Dellazizzo L, Potvin S, Giguère S, Dumais A. Evidence on the acute and residual neurocognitive effects of cannabis use in adolescents and adults: a systematic meta-review of meta-analyses. Addiction. 2022;117:1857–70.
-
Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophr Bull. 1987;13:261–76.
-
Mason OJ, Morgan CJM, Stefanovic A, Curran HV. The Psychotomimetic States Inventory (PSI): Measuring psychotic-type experiences from ketamine and cannabis. Schizophr Res. 2008;103:138–42.
-
Freeman D, Pugh K, Green C, Valmaggia L, Dunn G, Garety P. A measure of state persecutory ideation for experimental studies. J Nerv Ment Dis. 2007;195:781–4.
-
Spielberger CD, Gonzalez-Reigosa F, Martinez-Urrutia A, Natalicio LFS, Natalicio DS. The state-trait anxiety inventory. Revista Interamericana de Psicologia/Interamerican J Psychol. 1971;5:145–58.
-
Borgan F, Kokkinou M, Howes O. The cannabinoid CB1 receptor in schizophrenia. Biol Psychiatry Cogn Neurosci Neuroimaging. 2021;6:646–59.
-
Minichino A, Senior M, Brondino N, Zhang SH, Godwlewska BR, Burnet PWJ, et al. Measuring disturbance of the endocannabinoid system in psychosis: a systematic review and meta-analysis. JAMA Psychiatry. 2019;76:914–23.
-
Dempster EL, Wong CCY, Burrage J, Hannon E, Quattrone D, Trotta G, et al. Methylomic signature of current cannabis use in two first-episode psychosis cohorts. Mol Psychiatry. 2025;30:1277–86.
-
Whitehurst T, Howes O. The role of mitochondria in the pathophysiology of schizophrenia: a critical review of the evidence focusing on mitochondrial complex one. Neurosci Biobehav Rev. 2022;132:449–64.
Acknowledgements
We would like to thank the staff at the King’s Clinical Research Facility and Dr Luke Jelen for their support during the study. We sincerely thank BSPG Laboratories, IPS Pharma, and the Maudsley Hospital Pharmacy for providing and dispensing the study drugs. Without the collaboration of clinicians at the South London and Maudsley NHS Foundation Trust, recruitment for the study would not have been feasible. Finally, we would like to extend our heartfelt appreciation to our participants who generously volunteered their time and efforts to the study.
Funding
The study was funded by the National Institute for Health and Care Research (Doctoral Research Fellowship: NIHR300273).
Author information
Authors and Affiliations
Contributions
EC, PM, AE, DO, TPF and JS contributed to the design of the study. EC, AS, ADL, IS, ML, IGA, and NB recruited the participants. EC, AS, ADL, IS, ML, IGA, and DO conducted experiments and collected data. MW assisted with the sourcing and management of the study drugs. MK, AMD, MO analysed the plasma samples. DO and EC completed the statistical analyses. EC wrote the first draft of the manuscript. All authors reviewed and edited the final manuscript. All authors had full access to all the data in the study and had final responsibility for the decision to submit for publication. EC and DO have directly accessed and verified the underlying data reported in the manuscript.
Corresponding author
Ethics declarations
Competing interests
WL has provided consultancy for the clinical research organisation AxialBridge. AE has received speaker’s fees from Jazz/GW, Otsuka, and Lundbeck Pharmaceuticals. JS is a researcher and clinician who has worked with several pharma and tech companies to seek to identify new or improved interventions, but they do not have a relationship to the study and findings reported here. For further info see https://www.kcl.ac.uk/people/john-strang. All other authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Chesney, E., Oliver, D., Sarma, A. et al. Does cannabidiol reduce the adverse effects of cannabis in schizophrenia? A randomised, double-blind, cross-over trial.
Neuropsychopharmacol. (2025). https://doi.org/10.1038/s41386-025-02175-3
-
Received: 13 April 2025
-
Revised: 03 July 2025
-
Accepted: 08 July 2025
-
Published: 24 July 2025
-
DOI: https://doi.org/10.1038/s41386-025-02175-3
Search
RECENT PRESS RELEASES
Related Post




{kind=link}
{kind=link}
{kind=link}
{kind=link}